-
GEO生信数据挖掘(三)芯片探针ID与基因名映射处理
检索到目标数据集后,开始数据挖掘,本文以阿尔兹海默症数据集GSE1297为例
目录
上节我们下载了基因芯片平台文件并注释,我们发现存在一个芯片探针ID匹配到多个基因的情况,本节来介绍处理方案。
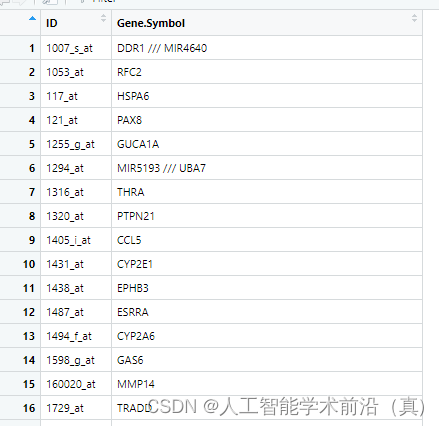
处理一个探针对应多个基因
我们通过简单检索发现两种方法:1.删除操作 2.保留分割符号前面的第一个基因
1.删除该行
- #处理一个探针对应多个基因
- #方案一:【删除该行】
- explan_final <- data.frame(explan_final[-grep("///",explan_final$"Gene.Symbol"),])
- #去一对多,grep是包含的意思,-就是不包含
2.保留分割符号前面的第一个基因
- #方案二:【保留第一个基因名】
- ids = platform_file_set #探针列名和基因名两列
- library(tidyverse)
- test_function <- apply(ids,
- 1,
- function(x){
- paste(x[1],
- str_split(x[2],'///',simplify=T),
- sep = "...")
- })
- x = tibble(unlist(test_function))
- colnames(x) <- "ttt"
- ids <- separate(x,ttt,c("ID","Gene.Symbol"),sep = "\\...")
- dim(ids) #探针列名和基因名两列
显然,第一个发现非常简单,在使用merge函数匹配时,会剔除更多的基因。第二个方法,会保留更多基因。
处理多个探针对应一个基因
表达矩阵中还有一个问题,如下图所示,很多探针指向同一个基因。

#把重复的Symbol 取每个基因所有探针的平均值或最大值作为基因的表达量
matrix <- aggregate(.~Gene.Symbol, matrix, mean) ##把重复的Symbol取平均值matrix <- aggregate(.~Gene.Symbol, matrix, max) ##把重复的Symbol取最大值
详细代码案例一删除法
- # 安装并加载GEOquery包
- library(GEOquery)
- # 指定GEO数据集的ID
- gse_id <- "GSE1297"
- # 使用getGEO函数获取数据集的基础信息
- gse_info <- getGEO(gse_id, destdir = ".", AnnotGPL = F ,getGPL = F) # Failed to download ./GPL96.soft.gz!
- # 提取基因表达矩阵
- expression_data <- exprs(gse_info[[1]])
- #查看平台文件列名
- colnames(annotation)
- #打印项目文件列表
- dir()
- # 读取芯片平台文件txt
- platform_file <- read.delim("GPL96-57554.txt", header = TRUE, sep = "\t", comment.char = "#")
- #查看平台文件列名
- colnames(platform_file)
- # 假设芯片平台文件中有两列,一列是探针ID,一列是基因名
- #probe_names <- platform_file$ID
- #gene_symbols <- platform_file$Gene.Symbol
- platform_file_set=platform_file[,c(1,11)]
- #将Matrix格式表达矩阵转换为data.frame格式
- exprSet <- data.frame(expression_data)
- #给表达矩阵新增加一列ID
- exprSet$ID <- rownames(exprSet) # 得到表达矩阵,行名为ID,需要转换,新增一列
- #矩阵表达文件和平台文件有相同列‘ID’,使用merge函数合并
- express <- merge(x = exprSet, y = platform_file_set, by.x = "ID")
- #删除探针ID列
- express$ID =NULL
- dim(express)
- exprSet = express
- #查看多少个基因重复了
- table(duplicated(exprSet$Gene.Symbol))
- #处理重复基因,计算行平均值方案1
- #rowMeans = apply(exprSet[,c(1:12)],1,function(x) mean(as.numeric(x), na.rm = T))####计算行平均值
- #处理重复基因,计算行平均值方案2
- #matrix <- aggregate(.~Gene.Symbol, matrix, mean) ##把重复的Symbol取平均值
- #row.names(matrix) <- matrix$Gene.Symbol #把行名命名为SYMBOL
- #处理重复基因,计算行平均值方案3
- library(limma) #avereps 函数
- exp_unique<-avereps(exp_symbol[,-c(32,ncol(exp_symbol))],ID=exp_symbol$Gene.Symbol)##把重复的Symbol取平均值
- #排序
- exprSet = exprSet[order(rowMeans, decreasing = T),]
- dim(exprSet)
- #去掉重复基因
- exprSet_2 = exprSet[!duplicated(exprSet[, dim(exprSet)[2]]),]
- dim(exprSet_2)
- #去掉缺失值
- exprSet_na = na.omit(exprSet_2)
- explan_final = exprSet_na[exprSet_na$Gene.Symbol != "",]
- dim(explan_final)
- #处理一个探针对应多个基因[删除法]
- explan_final <- data.frame(explan_final[-grep("///",explan_final$"Gene.Symbol"),]) #去一对多,grep是包含的意思,-就是不包含
- dim(explan_final)
- rownames(explan_final) <- explan_final$Gene.Symbol
- dim(explan_final)
- explan_final <- explan_final[,c(1:31)]
- # 此时explan_final为所需文件,行为基因,列为样本
> dim(explan_final)
[1] 12548 31详细代码案例二 多个基因名时保留第一个基因名
- # 安装并加载GEOquery包
- library(GEOquery)
- # 指定GEO数据集的ID
- gse_id <- "GSE1297"
- # 使用getGEO函数获取数据集的基础信息
- gse_info <- getGEO(gse_id, destdir = ".", AnnotGPL = F ,getGPL = F) # Failed to download ./GPL96.soft.gz!
- # 提取基因表达矩阵
- expression_data <- exprs(gse_info[[1]])
- # 提取注释信息
- annotation <- featureData(gse_info[[1]])
- #查看平台文件列名
- colnames(annotation)
- #打印项目文件列表
- dir()
- # 读取芯片平台文件txt
- platform_file <- read.delim("GPL96-57554.txt", header = TRUE, sep = "\t", comment.char = "#")
- #查看平台文件列名
- colnames(platform_file)
- # 假设芯片平台文件中有两列,一列是探针ID,一列是基因名
- #probe_names <- platform_file$ID
- #gene_symbols <- platform_file$Gene.Symbol
- platform_file_set=platform_file[,c(1,11)]
- #一个探针对应多个基因名,保留第一个基因名
- ids = platform_file_set
- library(tidyverse)
- test_function <- apply(ids,
- 1,
- function(x){
- paste(x[1],
- str_split(x[2],'///',simplify=T),
- sep = "...")
- })
- x = tibble(unlist(test_function))
- colnames(x) <- "ttt"
- ids <- separate(x,ttt,c("ID","Gene.Symbol"),sep = "\\...")
- dim(ids)
- #将Matrix格式表达矩阵转换为data.frame格式
- exprSet <- data.frame(expression_data)
- dim(exprSet)
- #给表达矩阵新增加一列ID
- exprSet$ID <- rownames(exprSet) # 得到表达矩阵,行名为ID,需要转换,新增一列
- dim(exprSet)
- #矩阵表达文件和平台文件有相同列‘ID’,使用merge函数合并
- express <- merge(x = exprSet, y = ids, by.x = "ID")
- #删除探针ID列
- express$ID =NULL
- dim(express)
- matrix = express
- dim(matrix)
- #查看多少个基因重复了
- table(duplicated(matrix$Gene.Symbol))
- #把重复的Symbol取平均值
- matrix <- aggregate(.~Gene.Symbol, matrix, mean) ##把重复的Symbol取平均值
- row.names(matrix) <- matrix$Gene.Symbol #把行名命名为SYMBOL
- dim(matrix)
- matrix_na = na.omit(matrix) #去掉缺失值
- dim(matrix_na)
- matrix_final = matrix_na[matrix_na$Gene.Symbol != "",]
- dim(matrix_final)
- matrix_final <- subset(matrix_final, select = -1) #删除Symbol列(一般是第一列)
- dim(matrix_final)
> dim(matrix_final)
[1] 14826 31小结
原始数据记录有22283条,多个探针对应一个基因采用取平均值处理,一个探针对应多个基因分别进行直接删除操作和保留第一个基因操作, 两种方法最终获得的数据记录分别为12548,14826。
更新版本的代码全文
- # 安装并加载GEOquery包
- library(GEOquery)
- # 指定GEO数据集的ID
- gse_id <- "GSE1297"
- # 使用getGEO函数获取数据集的基础信息
- gse_info <- getGEO(gse_id, destdir = ".", AnnotGPL = F ,getGPL = F) # Failed to download ./GPL96.soft.gz!
- # 提取基因表达矩阵
- expression_data <- exprs(gse_info[[1]])
- # 提取注释信息
- annotation <- featureData(gse_info[[1]])
- #查看平台文件列名
- colnames(annotation)
- #打印项目文件列表
- dir()
- # 读取芯片平台文件txt
- platform_file <- read.delim("GPL96-57554.txt", header = TRUE, sep = "\t", comment.char = "#")
- #查看平台文件列名
- colnames(platform_file)
- # 假设芯片平台文件中有两列,一列是探针ID,一列是基因名
- #probe_names <- platform_file$ID
- #gene_symbols <- platform_file$Gene.Symbol
- platform_file_set=platform_file[,c(1,11)]
- #一个探针对应多个基因名,保留第一个基因名
- ids = platform_file_set
- library(tidyverse)
- test_function <- apply(ids,
- 1,
- function(x){
- paste(x[1],
- str_split(x[2],'///',simplify=T),
- sep = "...")
- })
- x = tibble(unlist(test_function))
- colnames(x) <- "ttt"
- ids <- separate(x,ttt,c("ID","Gene.Symbol"),sep = "\\...")
- dim(ids)
- #将Matrix格式表达矩阵转换为data.frame格式
- exprSet <- data.frame(expression_data)
- dim(exprSet)
- #给表达矩阵新增加一列ID
- exprSet$ID <- rownames(exprSet) # 得到表达矩阵,行名为ID,需要转换,新增一列
- dim(exprSet)
- #矩阵表达文件和平台文件有相同列‘ID’,使用merge函数合并
- express <- merge(x = exprSet, y = ids, by.x = "ID")
- #删除探针ID列
- express$ID =NULL
- dim(express)
- matrix = express
- dim(matrix)
- #查看多少个基因重复了
- table(duplicated(matrix$Gene.Symbol))
- #把重复的Symbol取平均值
- matrix <- aggregate(.~Gene.Symbol, matrix, mean) ##把重复的Symbol取平均值
- row.names(matrix) <- matrix$Gene.Symbol #把行名命名为SYMBOL
- dim(matrix)
- matrix_na = na.omit(matrix) #去掉缺失值
- dim(matrix_na)
- matrix_final = matrix_na[matrix_na$Gene.Symbol != "",]
- dim(matrix_final)
- matrix_final <- subset(matrix_final, select = -1) #删除Symbol列(一般是第一列)
- dim(matrix_final)
- #+ 经过注释、探针名基因名处理、删除基因名为空值、删除缺失值 得到最终 matrix_final
- #+==================================================================================
- #+========================================================================================
已经完成了部分的预处理工作了,在使用数据前还有一系列的质控要做,请看下节数据清洗。
-
相关阅读:
Go 单元测试之mock接口测试
Acwing 828. 模拟栈
Python学习:if else对缩进的要求
Simple Factory Pattern 简单工厂模式简介与 C# 示例【创建型】【设计模式来了】
python实现外参标定的思路
【dp树状数组优化】跳跃(jump)
世界上到底有多少种编程语言
53. 最大子序和 392.判断子序列 115.不同的子序列 583. 两个字符串的删除操作 72. 编辑距离 编辑距离总结篇
【CSS】Flex布局及Quasar辅助类之Container
大数据技术之Hive:先导篇(一)
- 原文地址:https://blog.csdn.net/zzh1464501547/article/details/133303270
